












klaassen@mdc-berlin.de
Telefon: +4930450540656
Experimental & Clinical Research Center
Lindenberger Weg 80
13125 Berlin
Building 42-52, Room 1402

First, we clinically and genetically characterize different cardiomyopathy cohorts to identify novel disease genes, to understand the mechanisms for heart failure, and to improve prognosis. Second, we investigate the specific role of the transcriptional regulator PRDM16 for cardiomyocyte development, cardiac function, and cardiomyopathy. Third, we aim to assess how Bcl-2-associated athanogene 3 (BAG3) and the heat shock protein 70 (HSP70) complex affect cardiomyocyte proteostasis as a potential target for heart failure therapy.
Our concept bundles excellence in pediatric as well as adult cardiology, molecular mechanisms of inherited cardiac diseases, and therapeutic approaches in cardiomyopathy.
As a pediatric cardiogenetics group we focus on the molecular genetics of cardiomyopathies, arrhythmias, and structural congenital heart defects. Considerable progress in identifying the genes and pathways involved in left ventricular noncompaction cardiomyopathy (LVNC) has been made and this entity will continue be one main topic of our future research. Among the various projects and activities in the past 3 years at the ECRC the main progress has been made in identifying that mutation of the PRDM16 gene causes cardiomyopathy.
A short overview on our projects is shown in the corresponding sections.
1p36 deletion syndrome is recognized as the most terminal deletion syndrome. Here, we describe the loss of a gene within the deletion that is responsible for the cardiomyophathy associated with monosomy 1p36, and confirm its role in non-syndromic left ventricular noncompaction cardiomyopathy (LVNC) and dilated cardiomyopathy (DCM). Using our own data and publically available data from array comparative genomic hybridization (aCGH), we identified a minimal deletion for the cardiomyopathy associated with del1p36 that included only the terminal 14 exons of the transcription factor PRDM16 (PR domain containing 16), a gene that had previously been shown to direct brown fat determination and differentiation. Resequencing of PRDM16 in a cohort of 75 non-syndromic individuals with LVNC detected 3 mutations, including 1 truncation mutant, 1 frameshift null mutation, and a single missense mutant. In addition, in a series of cardiac biopsies from 131 individuals with DCM, we found 5 individuals with 4 previously unreported non-synonymous variants in the coding region of PRDM16. None of the PRDM16 mutations identified were observed in over 6400 controls. PRDM16 has not previously been associated with cardiac disease but is localized in the nuclei of cardiomyocytes throughout murine and human development and in the adult heart. Modeling of PRDM16haploinsufficiency and a human truncation mutant in zebrafish resulted in both contractile dysfunction and partial uncoupling of cardiomyocytes, and also revealed evidence of impaired cardiomyocyte proliferative capacity. In conclusion, mutation of PRDM16 causes the cardiomyopathy in 1p36 deletion syndrome as well as a proportion of non-syndromic LVNC and DCM.
Perspectives: Pathway analysis of PRDM16 shows that it is a very attractive candidate as a key regulator for cardiac hypertrophy and and remodeling. Data in zebrafish demonstrate that PDRM16 has as a dominant positive effect on cardiomyocyte proliferation showing that either activated or repressed levels of activity of PRDM16 impair proliferation. We will analyse the interaction partners of PRDM16 and its downstream targets.
Fig 1. LVNC, Echocardiography in Diastole
Fig 2. LVNC, Echocardiography in Systole
Fig 3. Mutations reside within the genomic sequence of exon 9 of PRDM16 in patients with LVNC
Fig 4. PRDM16 expression in the left ventricle of wild-type mice and human heart.
In adult human heart PRDM16 is expressed in the nuclei of both cardiomyocytes and interstitial cells (Fig. 4a). At mouse embryonic day (E) 13.5 (Fig. 4b), PRDM16 protein was expressed throughout the left ventricle but was most prominent in the endocardial and epicardial layers. In the adult mouse (Fig. 4c) PRDM16 expression was somewhat reduced overall compared to the embryonic heart but was still predominantly localized to the nuclei of cardiomyocytes. Taken together, these data support the hypothesis that PRDM16 is present and functional as a transcription factor in the nuclei of cardiomyocytes in the embryonic and adult mammalian left ventricular myocardium.
https://dzhk.de/aktuelles/news/artikel/neues-gen-fuer-herzinsuffizienz-entdeckt/
Fig. 1. Cardiac MRI of a Patient with LVNC
Left ventricular noncompaction cardiomyopathy (LVNC) is seen in a number of genetic syndromes and has been associated with neuromuscular disorders and with mitochondrial disease. It is classified as a distinct primary cardiomyopathy with a genetic etiology and may be an isolated finding or may also be associated with other forms of structural congenital heart disease (Jenni et al., 2001). Among many sporadic cases, familial recurrence with autosomal dominant inheritance is observed in isolated LVNC (Sasse-Klaassen et al., 2003).
The large number of mouse models that exhibit LVNC have been thought to represent a generic form of myocardial growth failure resulting from primary cardiac disease as well as from systemic growth disruption (Chen et al., 2009). There are discrete differentiation and maintenance programs for cardiomyocyte subpopulations within compact and trabecular myocardium (de la Pompa et al., 2012). LVNC has been proposed to result from either a reduction in the compact layer or a failure of maturation of the trabecular layer, but to date the primary mechanism remains ill-defined.
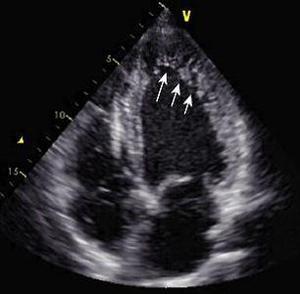
In humans, LVNC is morphologically characterized by a severely thickened two-layered myocardium consisting of a thin compacted epicardial layer and a thick noncompacted endocardial layer. The endocardial layer contains numerous prominent trabeculations and deep intertrabecular recesses (Fig.1, Fig.2).
We hypothesized that mutations in sarcomere protein genes may be associated with LVNC. In the first study (Klaassen et al., 2008) mutational analysis in a large cohort of 63 unrelated adult probands with LVNC and absence of other congenital heart anomalies was performed. Genes encoding 6 sarcomere proteins were studied. 11 mutations were identified in 3 distinct genes: β-myosin heavy chain (MYH7), α-cardiac actin (ACTC), and Troponin T (TNNT2). Nine distinct mutations, 7 of them in MYH7, 1 in ACTC, and 1 in TNNT2were found. LVNC is within the diverse spectrum of cardiac morphologies triggered by sarcomere protein gene defects.
In our second study we found that heterozygous mutations in genes encoding β-myosin heavy chain (MYH7), α-cardiac actin (ACTC1), cardiac troponin T (TNNT2),cardiac myosin-binding protein C (MYBPC3), and alpha-tropomyosin (TPM1) account for 30% of cases of isolated LVNC in adult patients (Probst et al., 2011). The distribution of disease genes confirms genetic heterogeneity and opens new perspectives in genetic testing in patients with LVNC and their relatives at high risk of inheriting the cardiomyopathy. The presence or absence of a sarcomere gene mutation in LVNC could not be related to the clinical phenotype.
In patients with LVNC, heart failure New York Heart Association class III or greater and cardiovascular complications at presentation were strong predictors for adverse outcome (Greutmann et al., 2012). In another study the aim was to find improved quantitative CMR criteria to distinguish LVNC from dilated cardiomyopathy (DCM) and hypertrophic cardiomyopathy (HCM) with high sensitivity and specificity. Absolute cardiac magnetic resonance (CMR) quantification of the total LV myocardial mass index (LVMMI) non-compacted or the percentage LV-MMnon-compacted and increased trabeculation in basal segments allows to reliably diagnose LVNC and to differentiate it from other cardiomyopathies (Grothoff et al., 2012).
Mutations in sarcomere protein genes in left ventricular noncompaction
Predictors of adverse outcome in adolescents and adults with isolated left ventricular noncompaction
20080604-mutationen_l_sen_schwere_herzmuskelerkrank
http://www.escardio.org/communities/Working-Groups/cmp/Documents/WGMP-Newsletter-Oct08-part-2.pdf
http://circ.ahajournals.org/content/117/22/2847.full.pdf
Fig. 2 Echocardiogram of a Patient with LVNC; arrows depict the noncompacted myocardium
Echocardiography (4-chamber view) of a patient with Ebstein anomaly and LVNC
A possible association between Ebstein anomaly with LVNC and mutations in MYH7 encoding ß-myosin heavy chain was tested in a large cohort of unrelated probands with Ebstein anomaly. Mutation-positive probands and family members showed various congenital heart malformations as well as LVNC. Significant pleiotropy and reduced penetrance were characteristic of MYH7 mutation-positive congenital heart malformations. MYH7 mutations were predominantly found in Ebstein anomaly associated with LVNC and may warrant genetic testing and family evaluation in this subset of patients. Ebstein anomaly is within the diverse spectrum of cardiac morphologies triggered by a sarcomere protein gene defect.Our study provides evidence for a link between structural proteins, cardiomyopathy, and congenital heart malformations (Postma et al., 2011). In adults, various forms of congenital heart disease are associated with LVNC, particularly stenotic lesions of the left ventricular outflow tract, Ebstein anomaly, and tetralogy of Fallot. In the future, studying these patients in more depth may provide a better understanding of the interplay between genetic and hemodynamic factors that lead to the phenotype of LVNC (Stähli et al., 2012).
Color doppler echocardiography of a patient with Ebstein anomaly and LVNC
Mutations in the sarcomere protein gene MYH7 in Ebstein's anomaly
In a number of projects the group is working on the molecular genetics of structural congenital hearts defects. The category of left ventricular outflow tract obstruction (LVOTO) is of major interest to us. Major collaboration partners here are R. Siebert (Director of the Medical Genetics Department, Kiel), HH Kramer (Director of the Pediatric Cardiology Department), Kiel, AA Arndt (Berlin/Boston) /C MacRae (Boston), and MP Hitz/ M Hurles (Wellcome Trust Sanger Institute, Cambridge, UK).
The studies are a comprehensive analysis of disease-associated changes using a panel of state-of-the-art technologies including whole-transcriptome analysis (RNAseq), copy number variation (CNV) analysis and whole-exome sequencing (WES).
In this collaborative study it was sought to determine the impact of structural genomic variation on left-sided congenital heart defects (CHD). In this first family-based CNV study of left-sided CHD, we found that both co-segregating and de novo events contribute to disease in a complex fashion at the structural genomic level. In our group we could confirm further that the CNVs in left-sided CHD were individual and rare.
Nonsyndromic atrioventricular septal defects (AVSDs) are an important subtype of CHDs for which the genetic architecture is poorly understood. We performed exome sequencing in 13 parent-offspring trios and 112 unrelated individuals with nonsyndromic AVSDs and identified five rare missense variants (two of which arose de novo) in the highly conserved gene NR2F2. We identified three additional CHD-affected families, among them individuals with LVOTO with other variants in NR2F2.
Rare copy number variants contribute to congenital left-sided heart disease
Rare variants in NR2F2 cause congenital heart defects in humans
http://www.kompetenznetz-ahf.de/presse/pressemitteilungen/erfolgreiche-suche-nach-einem-avsd-gen/
As part of the DZHK initiative (Deutsches Zentrum für Herz-Kreislauf-Forschung) the Pediatric Cardiology Department, DHZB (German Heart Centre Berlin), Director Prof. F. Berger, Berlin, participates for the partner-site Berlin. The general aim of the project is to characterize „Mechanisms of heart failure in congenital heart disease“. Genetic, molecular and imaging modalities are applied to identify predictors of adverse outcome.
Please contact us for further information.